Document Type : Review Article
Keywords
Subjects
Introduction
Blood and CSF biomarkers that were previously used to monitor the disease progression in the preclinical and clinical AD include tau, phospho-tau (p-tau), and Aβ42 (1-3). Since levels of tau and Aβ aggregates fluctuate as the disease progresses, understanding the relationship between the stage of disease and their levels is crucial is interpreting their relative significance to disease pathogenesis and clinical progression in AD. Accordingly, upregulation of p-tau and downregulation of Aβ in the blood/CSF was previously used as a reliable indicator for neurodegeneration in symptomatic AD patients (4). With synaptic loss occurring in parallel to neuronal damage, markers such as neurogranin and neurofilament (NFL) in CSF and blood were used to predict axonal instability and synaptic transmission disturbances in preclinical and clinical AD(5, 6). Visinin-like protein 1 (VILIP-1) which is a neuronal calcium sensor is also demonstrated to be in higher levels in the blood/CSF of AD patients as compared to control patients(7). Furthermore, measuring presynaptic and postsynaptic pro teins such as synaptosome-associated protein 25 (SNAP-25), and synaptotagmin in the blood/CSF yielded marginal success in estimating the neurodegeneration in the AD patients as compared to control patients (8, 9). Predicting neuroinflammation in AD can be successfully accomplished by assessing blood biomarkers such as MCP-1 and sTREM2
|
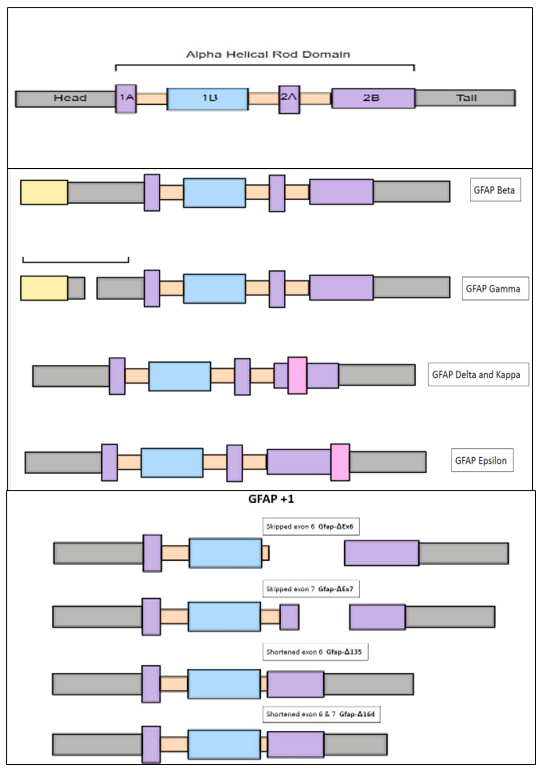
Figure 1.Structure of principal isoform of GFAP and alternate isoforms: The structure of principal isoform GFAP- α is composed of head, rod, and tail domain. GFAP-α, GFAP-β, GFAP-γ, GFAP-δ, GFAP-κ, and GFAP-ζ are the different isoforms that were widely expressed in the cortex, striatum, and cerebellum. These isoforms are derived by alternative splicing of GFAP- α. The pathological relevance of these uncommon GFAP isoforms in the context of AD disease pathogenesis is open to question and needs further investigation. But some of these isoforms were implicated in the pathogenesis of neurological diseases including Alexandria disease, astrocytoma, and glioblastoma. |
(10). Flotillin, which is an exosome marker was previously characterized for using it as a blood biomarker for gauging disease changes in AD(7). Previous reports established the fact that accumulation of Aβ aggregates in the brain tissue results in attenuated exosome secretion resulting in decreased serum flotillin levels (11, 12).
Positive emission tomography (PET) is an imaging modality that can utilize radioactive tracers for delivering crucial information regarding the shifts in the biochemical and metabolic activity of the brain tissues. PET has been used previously to ascertain and study Aβ aggregates, senile plaques, tau astrocytes, microglia, and synapses in AD with modest success(13-16). In this regard, the most studied PET tracer that targets MAO-B (mono amino oxidase-B) expressed in astrocytes for is 11C-deutrium-L-deprenyl (11C-DED)(17, 18). Tracers that are currently in the development for studying the subtle alterations in the levels of GFAP, tau and Aβ in the postmortem AD and control brain sections include 3H-PIB, 3H-THK5117 and 3H-florbetaben respectively(19-21).
GFAP: a future dependable marker for assessing disease-related brain tissue change in AD.
Any changes in the GFAP expression are deemed to suggest a disruption in the functional dynamics of astrocyte cytoskeleton, morphology, and their trophic protective function on neurons(22). This indirectly indicates that the onset of astrocytic response secondary to accumulation of Aβ and tau aggregates in AD(22). Indeed, astrocytes are deemed to be considered as a first line of brain immune cellular defensesthat are discharged upon appearance of Aβ aggregates in the neuronal tissues. Upon encountering the neuronal damage, young astrocytes tend to migrate near injured site and curtails further Aβ induced neuronal damage by their inherent secretory, phagocytic and trophic functions (21). As the inflammatory assault continues these young clusters of astrocytic populations will ultimately give up, but is slowly followed by a second wave of aged and resilient astrocytes with very high GFAP positivity thus increasing the chances of glial scar formation at the sites of neuronal injury(21). In parallel with these findings, a meta-analysis that examined the postmortem brain tissues concluded that there is a positive correlation between GFAP immunoreactivity and astrocytic response in AD(22). According to a three-center cross-sectional study conducted in Canada, plasma GFAP was a sensitive and reliable than CSF GFAP for assessing reactive astrogliosis and Aβ pathology in the early stages of AD (23). These findings are against the common logic where one would expect the CSF GFAP to better mirror unfolding neuronal damage in its vicinity rather than plasma GFAP which has crossed the blood-brain barrier into the systemic circulation.
Although plasma GFAP test is FDA-authorized for characterizing intracranial damage in traumatic brain injury (TBI), their potential usage in neurodegenerative and neuroinflammatory diseases warrants further validation. Studies examining the levels of GFAP in the CSF and blood conducted so far revealed encouraging albeit mixed and enigmatic results. This sets the precedence for reviewing the current literature on GFAP for its untapped potential as a CSF and blood biomarker in predicting the onset and progression of neurodegeneration in AD. Grasping, comprehending, and analyzing the current literature not only sets the stage for developing future basic science and clinical research studies for developing a reliable marker for diagnosing preclinical AD but also enables its potential usage as non-invasive marker for monitoring treatment in parallel with PET scan imaging modalities. A knowledge gap in understanding the relative importance of GFAP can be addressed a review that enlightens regarding its physiological structure, function, and current clinical studies involving GFAP in AD diagnosis and treatment monitoring
Given what has been said, this review was drafted to discusse systematically various topics ranging from GFAP structure, function, GFAP splice variants, physiological excretory pathways, post-translational modifications, importance of CSF and blood GFAP in relation to amyloid plaque deposition in AD. Another important aspect that needs further discussion is its head-to-head comparison with Aβ and phospho-tau levels to explore its sensitivity and specificity with and without these markers for predicting hippocampus atrophy in AD patients. Furthermore, it also be prudent to discuss whether GFAP monitoring needs to be supplanted with PET scans for earlier detection and risk stratification of AD. Taken together, the main purpose of this review is to update the current knowledge regarding GFAP so that it can kick-start future research studies to cement it position as one of the important and stable biomarker for early diagnosis as well as treatment monitoring in AD.
GFAP isoforms and their importance
There are several isoforms of GFAP reported to be expressed in the different brain regions. The underlying mechanism for generation of these different isoforms is through alternative splicing of GFAP mRNA(24). GFAP-α, GFAP-β, GFAP-γ, GFAP-δ, GFAP-κ, and GFAP-ζ are the different isoforms that were widely expressed in the cortex, striatum, and cerebellum(25, 26) (Fig 1). In AD mice, GFAP-α was most highly expressed isoform whereas other isoforms (GFAP-γ, GFAP-δ, GFAP-κ, and GFAP-ζ) were shown to have a variable expression with some reports establishing increased expression as compared to wild-type mice(25). Interestingly some prior studies show that, astrocytes which are positioned in proximity with amyloid plaques were stained positive for less common isoforms namely GFAP-α and GFAP-δ(25). The pathological relevance of these uncommon GFAP isoforms in the context of AD disease pathogenesis is open to question and needs further investigation.
Expression of some of these GFAP isoforms in the astrocytes leads to deleterious consequences due to their effect on filament assembly, function and cellular morphology. Expression of GFAP isoforms (GFAP-γ, GFAP-δ, GFAP-κ) in the astrocytes of AxD transgenic mice resulted in a spectrum of abnormalities ranging from attenuated filament solubility,
|
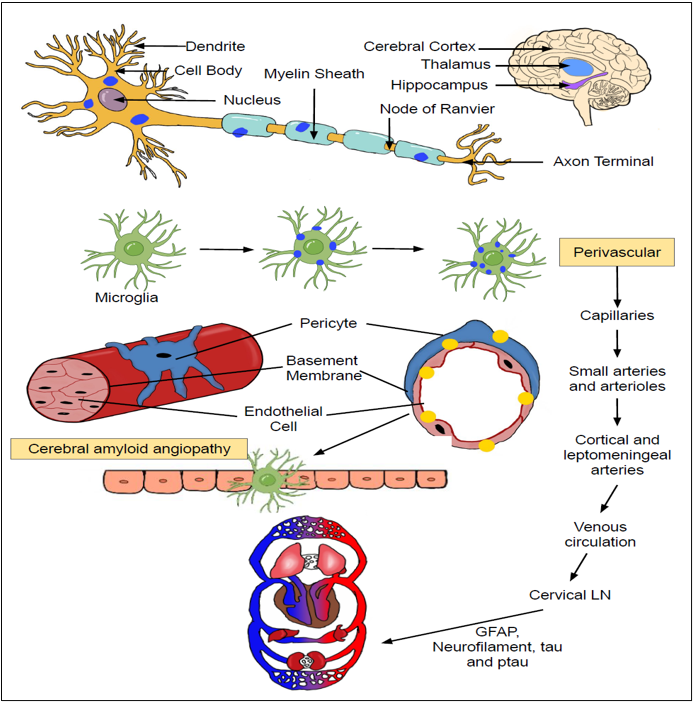
Figure 2. The pathological basis for emigration of GFAP and other toxic aggregates from brain circulation into blood: Amyloid deposits accumulating on the neuronal cells is primarily engulfed by scavenging microglial cells. These microglial cells then regurgitate these toxic aggregates upon the vascular endothelium of micro-vasculature thus triggering the onset of vasculitis and blood-brain barrier (BBB) dysfunction. The escape of GFAP from the brain circulation into the systemic circulation is dependent upon the presence of BBB dysfunction as well as its passage through the glymphatic and vascular pathways. The presence of an obstruction in these excretory clearance pathways can impede the efficient migration of GFAP in the systemic circulation, thus preventing it ability to be used as a biomarker for mirroring disease changes in AD. |
increased formation of cytoplasmic aggregates, derailed endogenous filament network, disordered astrocyte morphology, altered glutamate processing to disrupted trophic effect on the neighboring neurons(27). These findings exemplify the pathogenic influence of these GFAP isoforms in the pathogenesis of neurodegenerative diseases including AxD and AD.
GFAP-α:
Most of the literature published so far regarding the physiological and pathological functions of GFAP in the brain tissues is related to this predominant as well as principal isoform(28). This isoform has all the 9 canonical exons of a prototype GFAP gene(25, 28). Its protein length is approximately 432 amino acids (aa)(22, 28). In response to the neuronal injury, astrocytes respond with reactive gliosis which is characterized by morphological changes as well as increased expression of this main isoform(25). In sub-cellular mRNA localization studies, it was evident that most of the GFAP-α was localized to the astrocyte protrusions as compared to Gfapδ which can be attributed to subtle differences in 3'-exon sequences in their respective mRNA(24). Although this predominant isoform is responsible for supporting most of the astrocyte functions, any alterations in its physiological levels can trigger deleterious consequences.
Astrocytes isolated from transgenic mice overexpressing GFAP-α displayed a wide spectrum of abnormalities ranging from cytoplasmic inclusions, derailed filament network, attenuated proliferation, cell death to dysfunctional proteasomal system(29). High malignant grade (IV) astrocytoma is known to express low levels of GFAP-α and thus has higher GFAPδ/α ratio as compared low grade astrocytoma (II)(30). This change in GFAP isoform ratio might be detrimental as it can make astrocytes more motile, adhesive and promote their ability to invade the neighboring tissues.
This functional change in astrocytes might have some relevance to the behavior of astrocytomas. In this regard, gliomas having higher GFAPδ/α ratio were more invasive due to their increased cell adhesion and motility properties of astrocytes(31).
Forced overexpression of GFAP-α in the glioma cells lines resulted in decreased cell motility whereas its downregulation restored their motility and invasiveness(32). These findings underscore the importance of the GFAPδ/α ratio in controlling the expression of genes associated with cell proliferation, cell-ECM interactions, and protein phosphorylation in astrocytoma(30, 31). Using ex vivo brain slice model, it was shown that astrocytes with higher GFAPδ/α ratio displayed persistent invasion whereas those with lower GFAPδ/α ratio had disorganized migration(33). Previous studies demonstrated that GFAPδ/α ratio regulates physiological properties of astrocytes including cellular morphology, mitotic cycle extracellular matrix production, adhesion, 2D migration, phosphorylation, and proliferation(34). In a study a Moeton, M. et al, silencing of GFAP-α and increasing the GFAPδ/α ratio resulted in decreased motility via downregulation of plectin, increased ECM laminin and alteration of integrins(35). Therefore, it can be hypothesized that alteration of GFAP mRNA splicing and shifting the GFAPδ/α ratio in astrocytoma can be beneficial as it has the potential to impede the progression to highly malignant grade tumor, attenuate invasivesness, afford better clinical outcomes and prolong patient survival rates(30).
GFAP-δ:
GFAP-δ is distinguished from GFAP-α by the presence of an intron 7-8 sequences(25). Structurally, the exons 8 and 9 of GFAP-α isoform are replaced by exon 7a resulting in the generation of a new C-terminal tail domain(36). Due to incorporation of this C-terminal tail domain, it loses its ability to self-assemble into the intermediate filaments(37). As compared to GFAP-α, association and dissociation of GFAP-δ from the IF networks is also reported to be sluggish(38). This delayed filament dynamics can be explained due to tthe presence of coil-2B binding site in the C-terminal tail domain in contrast to GFAP-α(38). Nevertheless, GFAP-δ can still self-assemble in the physiological conditions when its threshold concentration is not breached (10%-15% of total GFAP concentration)(37). Incorporation of GFAP-δ into assembly compromised GFAP constructs (AxD GFAP mutant) results in deleterious effects such as increased protein aggregation, derailment of IF networks and increased association with αB-crystallin (protein chaperone) and p-JNK(37, 39). This disruption of IF networks results in alteration of astrocyte morphology with increased appearance of round cells with extended focal adhesions due to the shift in integrin and ECM interactions(38).
Overexpression of GFAP-δ tends to alter the cytoskeleton and filament assembly properties in a manner where there is a tendency towards intermediate filament collapse(37, 40). These findings highlight the fact that changes due to the incorporation of GFAP-δ might be due to the modification of filament-filament as well as filament-cytoplasmic protein interactions(37).
Astrocytes localized to the subpial zone of the cerebral cortex, spinal cord, olfactory bulb, human rostral migratory pathway, sub-granular zone of the hippocampus and along the ependymal layer of the cerebral ventricles express this GFAP-δ isoform(37, 40, 41). GFAP-δ positive astrocytes are found most abundantly in the sub-ventricular zone (SVZ) or along lateral ventricles, where they function as multipotent neuronal stem cells(40, 42, 43). Although both GFAP-δ and GFAP-α were documented to be upregulated in the AD lesions, GFAP-δ upregulation only transpires via alternative splicing from GFAP-α isoform in the likely setting of enhanced GFAP gene expression(40, 44). Other than AD, its expression is documented in pathologies such as astrocytoma, vanishing white matter syndrome, and stroke(39, 45, 46).
According to Heo, H.D. et al, the intensity of GFAP-δ staining is directly proportional to the grade of spinal cord astrocytoma and thus it has the potential to become a reliable marker for monitoring the histological grade, tumor behavior and invasiveness(45).
Higher GFAPδ/α ratio leads to poor prognosis in gliomas as it promotes increased astrocyte-ECM interactions via DUSP4 (Dual specificity phosphatase 4)(31). DUSP4 which regulates MAPK pathway has a role in a variety of biological processes including migration, invasion, proliferation, ECM degradation and chemotherapy-induced cytotoxicity(36). Alternatively, higher GFAPδ/α ratio leads to increased invasiveness in the malignancies through laminin mediated upregulation of phospholipase-D, phosphatidic acid, and matrix metalloproteinase-2 (MMP-2)(35, 36, 47, 48). MMP2 which is a type IV collagenase is utilized by glioma cells to invade across ECM and thus its overexpression increased the likelihood of metastasis and poor survival in the gliomas(36, 47, 49).
|
Figure 3. Arterial phase of GFAP excretory pathway. The peri-vascular excretory pathways of GFAP are powered by peri-arterial pulsations, sleep and respiration. This pathway is briefly classified into arterial phase, capillary phase, and venous phase. In the arterial phase, accumulated solutes and GFAP floating in the extracellular space will eventually travel closely along the endothelial basement membrane and vascular smooth muscle of arterial vasculature. The aquaporin channel (AQP4) which is present on the astrocytic end feet and very closely juxtaposed to the vascular endothelial basement membrane. This AQP4 channel will be responsible for filtering and secretion of these solutes including GFAP from the peri-arterial space into the brain interstitial fluid by peri-arterial efflux. |
GFAP+1
This isoform comprises of four variants namely GFAPΔEx6 (exon-6 of GFAP-α deleted), GFAPΔEx7 (exon-7 of GFAP-α deleted), GFAPΔ164 (abbreviated exon-6&7 of GFAP-α deleted along with absent Coil 2B), and GFAPΔ135 (abbreviated exon-6 of GFAP-α deleted along with absent Coil 2B)(22, 25, 26, 28). GFAP+1 positive astrocytes were identified with purified GFAP+1 positive antibody in the sub-ventricular region, hippocampus, striatum, spinal cord of the elderly non-demented controls, AD and Parkinsonism (PD) patients(44, 50). Recent reports were successful in identifying these out of frame splice variants in the pyramidal neurons of hippocampus in the AD and down syndrome patients(51). The researchers in study speculate that the appearance of these splice variants can be reasoned by occurrence of increased GFAP gene stimulation along with impairment of proteosome degradation(51). A previous study correlated the presence of GFAP+1 positive astrocytes in the hippocampus to the accumulation of Aβ1–42 oligomers and fibrils in the AD patients(44). Alternative mechanisms hypothesized for the appearance of these isoforms include somatic mutations, genetic modification, DNA mutations and stable epigenetic alterations with increasing age (44, 52). GFAP+1 was also highly expressed in the focal brain lesions of chronic epilepsy associated with astrogliosis (53). The immunostaining pattern of GFAP+1 positive astrocytes appear to extend 1-5 long cellular processes (1 mm) that can be traced to nearby blood vessels or neutrophils in contrast to GFAPδ positive astrocytes which appear as stained cellular bodies with predominant peri-nuclear pattern (28, 40, 50, 53, 54). Another distinguishing feature of GFAP+1 positive astrocytes is the absence of evenly positioned varicosities on the long cellular processes emanating from these astrocytes (55). Transfection of GFAP+1 isoform into the SW-13cl.2 cells which lack the endogenous expression of intermediate filament proteins resulted in the collapse of the intermediate filament network along with altered filament-filament interactions thereby underscoring its pathological role in hampering normal filament-filament coordination (44). Furthermore, the predominance of this isoform over canonical GFAPα in the astrocytes accelerates the wreckage of IF network leading to disfigured astrocyte morphology(44).The impact of GFAP+1 isoform on the filament assembly, cellular morphology and physiological function needs further clarification and research in the near future.
GFAPγ
This GFAP isoform is formed by deleting exon 1 and replacing it with intron 1-2(25, 56). In mice, it is expressed in the astrocytes, bone marrow and spleen whereas in humans it is only detected in the brain(56). Due to lack of amino-terminal head domain, it might not be ablw to function as intermediate filament (56). The significance of its expression and its primary physiological function in the astrocytes needs further validation.
GFAPκ
This isoform can be differentiated from GFAP-α by the presence of exon 7b formed by a linking of sequences including exon 7, intron 7a and exon 7a(57). It is produced by combination of events namely alternative splicing and polyadenylation(57). Its expression is documented in the adult human frontal cortex, human glioblastoma tumors and human astrocytoma cell lines (U343)(57, 58). GFAPκ is less capable to form homomeric filaments and this compromised filament assembly can be attributed due to the absence of RDF motifs in the C-terminal tail and positive charge of the tail domains(59, 60). In fact, co-expression of GFAPκ and GFAPα resulted in the formation of an unstable IF network and protein aggregates(57). Previous reports indicate that GFAPκ/GFAPε ratio is a marker for glial cell differentiation and any increase in this ratio signifies the aggressiveness of glioblastoma tumors(57). This can be reasoned by the hypothesis that GFAPκ fine-tunes the IF filament assembly and revamps the shape in a manner that makes the tumor cells more motile, adhesive and invasive. These changes might increase the chances of developing aggressive tumors, and resultant worse prognosis and poor clinical outcomes(57). Regardless, the influence of this isoform on the IF assembly, network and associated cellular functions in the physiological and pathological states needs further attention. Furthermore, it would also be worthwhile to investigate the impact of this isoform on the disease progression as well as neurodegeneration in AD.
GFAPβ
GFAPβ is identified by the presence of secondary upstream transcription site at the classical 5’ UTR region normally present in the canonical GFAPα isoform(25, 61). Northern blot analysis confirmed the expression of this isoform in the primary astroglial cultures(61, 62). It is also isolated from rat schwannoma cell lines, rat cerebral cortex, hamster brain and human brain(62). Neurotoxic brain injury in the rat hippocampus was documented to stimulate the expression of both isoforms GFAPα & GFAPβ within the astrocytes(61). These findings underscore the existence of analogous post-transcriptional mechanisms in place for modulating the expression of these isoforms in response to neural injury(61). Treatment of rat astrocyte cultures with 25U of Interferon (IFN) γ resulted in increase in the GFAPβ levels along with small decrease in the total GFAP levels(62). Under normal conditions, this isoform accounts for at least 5-10% of total GFAP(62). In rat brain slice cultures Treatment of rat brain cultures with 25U of IFN-γ did not have any effect on the expression of GFAPβ levels while decreasing the total GFAP levels by 10-fold, thereby increasing its relative concentration to 50%(62). According to Benvenisate et al, actions of IFN-γ can be explained by stimulation of protein kinase C (PKC) in the astrocytes of rat brain slice cultures(63). This relative upregulation of this isoform with IFN-γ might represent a compensatory response for activating different set of astrocyte populations to defend against the inflammatory stimulus(62). Further studies should be focused on understanding the transcriptional regulation of this GFAPβ isoform in physiological and pathological conditions.
GFAPζ
This isoform is characterized by the presence of 284 bp intron 8-9 interspersed within the GFAP gene(25). Its physiological and pathological role in the astrocytes and brain tissues is currently unknown. The importance of this isoform and its relative effect on the filament assembly, dynamics and functional aspects needs further elucidation.
Mechanisms proposed for dribbling of GFAP from cerebrospinal fluid into the systemic circulation:
Astrocytes are the primary glial cells that respond to TBI or neuronal injury secondary to accumulation of toxic Aβ and tau plaques (64-66). Reactive astrogliosis is a term highlighting the morphological changes in the astrocytes for mounting a robust immune response to contain and thwart any neurological damage secondary to inflammatory or toxic stimulus (67). Reactive astrogliosis can encompass a mosaic of changes ranging from alteration of gene expression, change in the morphology, and cytoskeleton hypertrophy, to upregulation of GFAP(67). It is important to understand that the degree of GFAP upregulation is proportional to the degree of CNS tissue damage in disorders such as neurodegenerative diseases, inflammation, infection, stroke and autoimmune diseases(67).
Unfortunately, astrocyte damage and cell death can be a by-product of moderate to severe astrogliosis, and this will result in spilling of GFAP from astrocytes into the brain interstitial fluid (ISF) and cerebrospinal fluid (CSF)(67, 68). These upregulated levels of GFAP in the brain interstitial fluids can be a reliable signature that can signify the severity of reactive astrogliosis but it might not show a direct relationship with the severity of neurodegeneration in dementia patients(69). Once GFAP enters the CSF, it circulates and courses along the vascular channels and eventually decamps from the brain into the systemic circulation secondary to structural abnormalities or due to efficient excretory systems in place.
GFAP circulates in the ISF, and CSF makes it way towards the systemic circulation either due to BBB dysfunction or via seeping through brain’s glymphatic system or meningeal lymphatic vessels(70-74). In the following sub-sections, each of these excretory pathways utilized for GFAP will be discussed in detail.
Blood-brain barrier disruption:
Traumatic brain injury can disrupt the BBB and allow the seepage of albumin, fibrinogen, and thrombin from the systemic circulation into the brain(70). These factors will ultimately stimulate astrocytes and microglia adjoining the vascular endothelium of BBB leading to secretion of reactive oxygen species (ROS), Nitric oxide (NO), tumor necrosis factor-alpha (TNF-α), IL-1 β, IL-6, IL-12, glutamate, chemokines, and MMPs(70). These factors will cause apoptosis of the vascular endothelial cells and derail the integrity of BBB(70). This will allow more inflammatory cells seepage into the brain to further exacerbate the BBB damage already initiated as well as instigate post-traumatic neuroinflammation(70). In AD, a different mechanism of BBB disruption has been proposed. Accumulation of toxic Aβ aggregates seems to trigger activation of vascular endothelium. Energized vascular endothelium then upregulates adhesion molecules like ICAM-1 (Intracellular adhesion molecule-1) & VCAM-1 (Vascular cell adhesion molecule-1) and chemo-attractants which then sparks the immigration of neutrophils from the systemic circulation into the brain(75, 76). These drifted neutrophils will be activated by surrounding Aβ & senile plaques thereby acquiring a virulent phenotype(77). These stimulated neutrophils will secrete a spectrum of toxic factors including ROS, cytokines, chemokines, and neutrophils extracellular traps (NETs)(76, 78). These pro-inflammatory agents are alleged to trigger BBB dysfunction, chronic inflammation, reciprocal microglial activation, neuronal degeneration, and synaptic dysfunction(76). Furthermore, oligomeric Aβ accumulated within the neurons and synapses will be ingested and shuttled by neighboring microglial cells to the endothelium of microvasculature thereby instigating the onset of cerebral amyloid angiopathy (CAA)(79)(Fig 2). Initiation of CAA on the cerebral microvasculature by deposition of amyloid plaques will cause the breakdown of BBB and the subsequent structural abnormalities would spark the leakage of GFAP from brain interstitial fluids into the systemic circulation.
During CAA, Aβ accumulation starts initially on the basement membrane and progresses gradually inwards to replace the entire arterial wall (Tunica interna, Tunica media and Tunica externa)(80)(Fig 2). Studies demonstrate that this occurrence of CAA varies among different brain regions with increasing age and AD(81). In mouse models of AD, hippocampus, thalamus, and cerebral cortex exhibit higher levels of CAA whereas striatum is least effected in terms of vessel wall changes(81). This CAA can itself contribute to BBB dysfunction via decreased expression & mis-localization of tight junction proteins (Claudin-5 & Zonula occluden-2), and increased expression of MMPs(82-85). Furthermore, it has been documented that amyloid beta is directly translocated across the BBB utilizing receptors LDL receptor-related protein-1 (LRP-1) and alpha (2)-macroglobulin(86). ATP binding cassette (ABC) transporters such as ABCB1 (also known as P-glycoprotein 1 or MDR1) is capable to transporting Aβ across the BBB into the systemic circulation(87). Internalization and elimination of Aβ across the brain capillary endothelial cells at BBB is enforced by insulin and human natriuretic peptide (hANP) via engagement of insulin degrading enzyme (IDE)(88). It is quite possible these receptors in BBB will be damaged and dysfunctional in AD patients due to continuous onslaught by these toxic inflammatory mediators secondary to profuse accumulation of Aβ aggregates within the brain tissues(86, 89-91).
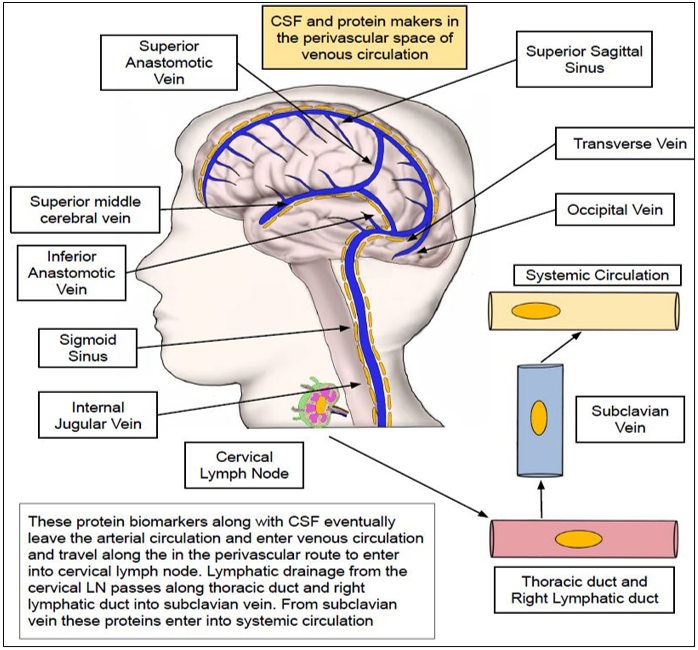
Perivascular route and Glymphatic clearance pathways:
Markers such as GFAP, tau, synuclein, neurofilament and amyloid beta can passively slide through along the basement membrane of the smooth muscle of microvasculature in a peri-vascular route and drain into the cervical lymph nodes (LNs)(81, 82, 89, 92, 93). This peri-vascular route is energized by the low frequency peri-arterial pulsations and spontaneous vasomotion which was recently demonstrated with in-vivo two-photon microscopy by van Veluw SJ et al(94, 95). Additionally, respiration and CSF pressure gradients were known to play atleast a partial role in rapid inter-exchange of fluids between CSF and ISF(96, 97). Other physiological factors that are hypothesized to play a significant role in the operation of perivascular and glymphatic clearance pathways include APOE*ε4, arterial age, molecular size /weight of solutes, AQP4 expression and sleep(98).
It has been shown that sleep directly accounted for 40% clearance of Aβ aggregates via the glymphatic clearance pathway during physiological conditions(98). This is because there is 60% increase in the interstitial space during sleep resulting in the exuberant exchange between CSF-ISF compartments promoting the Aβ clearance via lymphatic pathways(98, 99). Continuous cardiac output is essential for peri-vascular drainage of soluble solutes and fluid without any cells via capillary and arterial basement membrane which function as surrogate lymphatics of the brain(100).
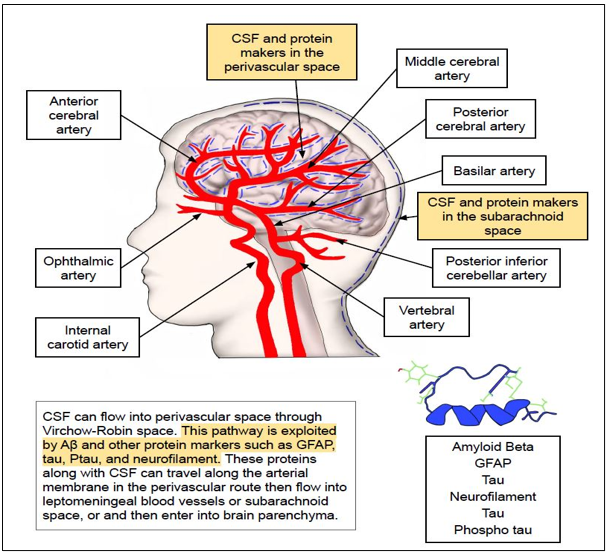
Broadly, this glymphatic pathway can be divided into five phases namely peri-arterial influx, peri-arterial efflux, CSF-ISF exchange in the brain parenchyma, peri-venous influx, and peri-venous efflux(101-103)(Fig 3-5).
In this pathway, accumulated extracellular solutes in the brain that need to be cleared from CSF circulation initially enter an arterial peri-vascular space where they travel closely abutting along the endothelial basement membrane and vascular smooth muscle (peri-arterial influx) (Fig 3) (82, 96-98, 101). Preston et al, argued that there is a slightly modified perivascular drainage pathway whether solutes from the extracellular surface of the brain initially move into the peri-capillary spaces and later into the peri-arterial space of cortical and leptomeningeal arteries(89). From peri-arterial space,
|
Figure 4. Capillary phase of GFAP excretory pathway. In the capillary phase, the solutes secreted will traverse through the capillary network of the arterial and venous vessels. In this phase, there will be inter-mixture of solutes secreted from the cerebrospinal fluid (CSF) and brain interstitial fluid and they travel across the brain parenchyma. This phase precedes the onset of venous phase from where these solutes will be finally making their way into the systemic circulation by squeezing through the lymphatic vessels and lymph nodes. |
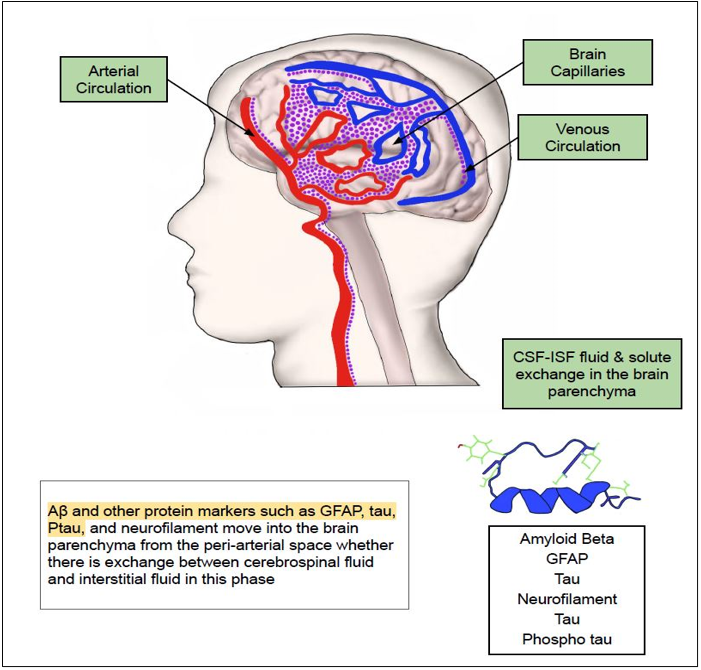
these solutes are then secreted by AQP4 (Aquaporin-4) channel present on the astrocytic end feet which is closely juxtaposed to the arterial endothelial basement membrane into the ISF (peri-arterial efflux) (82, 96-98, 101). Downregulation of astrocyte water transport by blocking the expression of AQP4 resulted in 70% reduction in CSF influx into the brain interstitial space and 40% attenuated interstitial solute drainage via the glymphatic system(101, 104, 105). Next, these secreted solutes move across the brain parenchyma where is an intermixture of solutes from CSF & ISF by convection (Fig 4). In the subsequent phase, they enter the perivascular space of venous circulation closely bordering the endothelial basement membrane (peri-venous influx) (82, 96-98) (Fig 5).
By intrathecal administration of contrast agents, researchers were able to visualize the peri-arterial CSF influx and CSF-ISF fluid changes in the extracellular space of the rat brain with the help of dynamic contrast-enhanced MRI(106). By sliding across venous circulation these solutes are eventually transported to the cervical LNs by (peri-venous efflux). Once they enter the lymphatic circulation, these are emptied into the right lymphatic duct and thoracic duct. From the lymphatics, these solutes are drained into the sub-clavian vein and finally into the systemic arterial circulation.
Implications for impaired peri-vascular drainage:
Any impedance of this peri-vascular drainage is detrimental as it facilitates the accumulation of these toxic aggregates within the brain parenchyma thus raising the chances of accelerated neuronal death as well as neuroinflammation in neurodegenerative diseases (Fig 2). Mice overexpressed with apolipoprotein E (APOE) ε4 allele developed profuse accumulation of Aβ(40) in the hippocampus secondary to blockage of peri-arterial drainage pathways via collagen deposition in vascular endothelial basement membrane as compared to wild-type mice(107). According to Nimmo et al, age-related decline in the peri-arterial drainage pathways can be the underlying mechanism for the manifestation of various synucleinopathies and tauopathies(108). Research studies indicate that the most important factor governing the efficacy of peri-vascular drainage in aged mice is the presence of CAA(95, 109). The degree of peri-vascular drainage varies across different brain regions, and this can be partly explained by disparities in the intensity of CAA present in their respective regional vasculature(110). Failure of peri-vascular drainage due to CAA can ultimately result in the rupture of the Aβ laden arteries (Intra-cerebral hemorrhage), deposition of Aβ in the brain tissues, disarray of brain homeostasis, which in combination incites neurodegeneration in AD(95).
Significance of CSF GFAP in amyloid plaque deposition in pre-clinical and clinical AD.
Assessing and characterizing CSF biomarkers including GFAP for neurodegenerative diseases is important because their changes are commensurate with and might provide indirect evidence of ongoing pathological processes in the brain including protein misfolding, protein aggregation, astrogliosis, microglial overactivity, synaptic dysfunction and neuronal death(111). Immunological determination of CSF GFAP levels by Fukuyama et al in 2001 revealed that their levels are higher in the moderate to severe AD patient cohorts (13.2 ± 9.10 ng/ml) as compared to mild to moderate AD groups (6.85 ± 5.76 ng/ml)(112). Jesse S, et al that appraised the CSF levels found that GFAP levels in the AD patients were significantly higher than Creutzfeldt-Jakob disease (CJD), and non-demented control patients (CON), underscoring its fluctuation with neuronal tissue damage in neurodegenerative diseases(113). Accordingly, CSF levels of GFAP, YKL-40 (chitinase-3-like protein 1), CHIT1 (chitotriosidase-1) were elevated in the prion disease, AD, and frontotemporal dementia (FTD), as compared to controls(114). Nevertheless, none of them except YKL-40 performed better in terms of sensitivity and specificity for offering a reliable diagnostic value for differentiating high risk groups from controls(114).
In multiple sclerosis (MS), CSF GFAP levels are higher than controls, with its values more unequivocal and dependable for progressive MS as compared to relapsing-remitting MS for forecasting disease evolution, prognosis, and patient survival(115). In MS patients, elevated GFAP is a marker of pathological changes including severe astrogliosis, and irreversible neurological damage, a combination that might be perceivable as a harbinger for future neurological disability in these patients(111, 116, 117). In a study performed by Jany, P.L. et al analyzing the GFAP levels with ELISA immunoassay in 24 patients in AxD, CSF GFAP levels were significantly and consistently elevated as compared to controls in contrast to blood GFAP levels which were only upregulated in a few subsets of patients(118). AxD patients have underlying astrocytic injury, seizures, hydrocephalus, and cavitation which might inherently amplify the spillage of GFAP from astrocytes into the extracellular space and CSF within the brain(118).
Not to mention, AQP4 water channel of the astrocytic end feet might be disintegrated in these AxD patients thus forestalling the exuberant clearance of this extracellular GFAP floating around in the brain ISF through the peri-vascular and glymphatic pathways into the systemic circulation(118, 119). In a recent observational cross-sectional study where they prudently planned to gauge the blood and CSF biomarkers from 3 centers from July 2014 to January 2020, CSF GFAP was overmatched by blood GFAP in accurately portraying the Aβ pathology in different AD continuum groups(23). The authors speculate that, CSF GFAP provides a more acceptable mirror image of the astrocytic counterattack in response to microglial activation instead ofencumbrance in the AD continuum groups(23). Moreover, augmented spilling of the GFAP from CSF into the systemic circulation secondary to blood brain barrier dysfunction and peri-vascular drainage is the most probable explanation for under-performance of CSF GFAP in relaying Aβ pathology in AD patients (23). In parallel with these findings, in a study completed by Montoliu-Gaya L et al, CSF GFAP levels demonstrated insignificant fold change and were found to be a second fiddle for predicting pre-symptomatic as well as symptomatic AD with respect to cortical thinning and amyloid Aβ uptake (120). Additionally in a clinical trial recently performed by Pereira, J.B. et al, CSF GFAP underperformed as a biomarker as it did not correlate with hippocampal Aβ accumulation, cognitive impairment, and PET findings in different AD groups (121). Definitively, it exhibited a lesser precipitous upsurge with Aβ burden on PET scans and was also increased in non-AD neurodegenerative disorders(121). Regrettably, even after combining with other markers such as sTREM2 and YKL-40 the performance of CSF GFAP as a marker for inferring Aβ burden on PET scan was very displeasing and unfavorable (121). In contrast in a clinical research study performed by FerrariS. J.P. et al, CSF GFAP were accurate enough to accurately predict Aβ conglomeration in the PET scan as compared to CSF YKL-40 which presaged PET tau agglomeration in the AD patients(122). These divergent findings draw attention to the fact that CSF GFAP elevation arising from astrogliosis can be inferred as non-credible and non-specific. Furthermore itselevation can surface with any neuroinflammatory or neurodegenerative clinical condition without any clear-cut attribution to Aβ stock-piling specifically associated with AD patients.
|
Figure 5. Venous phase of GFAP excretory pathway. In this phase, solutes enter the peri-vascular phase of the venous circulation where they course very closely to the endothelial basement membrane of the venous vessels. Eventually, from the venous vessels they will enter the lymphatic circulation and are ultimately emptied into the right lymphatic duct and thoracic duct. From here, the solutes are emptied into the sub-clavian vein and thereby finally emigrating into the systemic circulation. |
Why CSF GFAP underperforms as a biomarker in predicting structural abnormalities and Aβ accumulation in pre-clinical and clinical AD groups as compared to blood GFAP
From the above discussion, it is obvious that there some conflicting opinions regarding the suitability of CSF GFAP as upright and unequivocal marker for guessing the underlying Aβ pathology in the preclinical and clinical AD groups. So, few researchers contended some speculative hypothesis and opined plausible guesswork for decreased stability as well as enhanced degradation of GFAP in the CSF thus making it a less viable option for monitoring disease progression-related brain changes in AD.
First, freeze thaw cycles and storage temperature might have a varying effect on the stability of GFAP in the blood and CSF samples. In a study where paired blood and CSF samples from AD patients went through a series of freeze-thaw-cycles (FTC) followed by GFAP assessment by molecule array technology, CSF GFAP levels declined by about 188.12 pg/ml per each FTC while blood GFAP levels were untarnished due to their higher endurance capacity(123). Blood GFAP levels endured repeated four freeze-thaw cycles and were more resilient from degradation as compared to Aβ40 and p-tau181 levels which underwent degradation with more than three freeze-thaw cycles(124). It was previously demonstrated that GFAP is cleaved by calpain and caspases into BDPs including rod domain as well as N&C terminal fragments(22, 125). ELISA anti-GFAP antibodies were previously shown to detect the GFAP and its fragments in the CSF and blood samples within the molecular weight range 38-50 kDa(126). It is conceivable that pattern of GFAP cleavage by calpain and caspases is quite inconsistent as well as unequable in CSF and blood samples. Due to this erratic GFAP cleavage, generated divergent GFAP fragments might have contrasting molecular weights (20 kDa) in these respective body fluids (127). This might make the GFAP fragments (20kDa) evading discovery by routinely used in ELISA detection kits for being far-flung from the detection range of anti-GFAP antibodies routinely used by them(126). This might partly account for differences in detecting GFAP levels in the CSF and blood. Moreover, it has been estimated that the amount of time taken by CSF GFAP to traverse the cerebral vasculature to reach the lumbar sac is approximately 4h(128). It is quite possible that GFAP might have been partially degraded is the CSF by the time it reaches its final destination (lumbar sac).
Research studies should be focused on the direction of accurately accounting for the different GFAP fragments generated in the CSF / blood and appraising their specific molecular weights. This will enable the researchers to design and tailor specific GFAP fragment specific antibodies which can accurately track down these spliced breakdown products. This will ultimately uncover reliable biomarkers capable of ferrying out disease progression in AD continuum groups.
Second, GFAP released from the damaged astrocytes will initially take refuge in the CSF and brain interstitial fluids. This GFAP retreat in the CSF might be abbreviated by its high turnover rate secondary to its massive degradation rate and enhanced seepage pathways into the systemic circulation. Lower levels of CSF GFAP levels as compared to blood can be partially explained by the operation of adeptdeporting mechanisms through peri-vascular route and glymphatic pathways(129). Furthermore, BBB dysfunction which frequently accompanies AD patients also facilitates augmented spillage of GFAP from CSF into the systemic circulation(130-132). Thus, once GFAP is released extracellularly from astrocyte damage, it might quickly escapes from CSF into systemic circulation through the above-mentioned channels.
Previous reports state that phosphorylation of GFAP kindles depolymerization of filamentous subunits and affords buffering from enhanced proteolysis in the body fluids including blood and CSF(133). It is probable that the ratio of phosphorylation-dephosphorylation of GFAP in the CSF is doctored so that dephosphorylation supersedes thus yielding towards augmented proteolysis as well as enhanced degradation of GFAP in the CSF. On the contrary, dephosphorylation might take a back seat allowing the phosphorylation to overshadow in the blood thereby bolstering the filament integrity and structural plasticity of the GFAP filaments. These changes in the filament assembly due to post-translational modification of GFAP might partly explain the discrepancy in the GFAP levels in the CSF and blood.
Although these are some of the speculations for the mechanisms that can conceivably happen behind the curtain for exaggerated degradation, as well as amplified spurting of GFAP from CSF, the actual reasons are still enigmatic and cryptic. The need of the hour is launching basic science and clinical research studies to comprehend these unsolved mysteries. This will ultimately lay the appropriate foundation for shedding the light on GFAP stability, turnover, phosphorylation status and excretory pathways. Discerning this crucial information will enhance the practical utility of CSF GFAP as a reliable biomarker for predicting onset and clinical progression of disease pathology in AD.
Upper hand of serum GFAP as a biomarker in predicting structural abnormalities and Aβ accumulation in pre-clinical and clinical AD groups
A wide variety of clinical research studies conducted so far underscore the importance of blood GFAP as a crucial biomarker that can appraise and capture the structural brain abnormalities, disease evolution and disease progression in AD. Currently guidelines suggest that it can be used as screening as well as disease monitoring tool for making informed treatment decisions in AD when used carefully in tandem with clinical assessment and imaging modalities. In this section, we will summarize the clinical research studies that highlight its potency as biomarker in gauging clinical progression in AD.
Blood GFAP was shown to predict the excessive accumulation of Aβ42/40 and Aβ42/T-tau in the brain tissues in patients with AD pathology(134). In addition to that it was also demonstrated to be efficient in predicting the evolution of this high-risk cohort into overt AD with mild cognitive impairment (MCI)(134).
According to a study by Ranjan, K.B. et al, patients with baseline blood GFAP values > 232 pg/ml of GFAP exhibited a 130% greater reduction in hippocampal volume as compared to those with baseline values less than 160 pg/ml(135). In the analysis of blood samples from 300 patients from Amsterdam Dementia Cohort which were studied for 3 years, both blood GFAP and amyloid β42/40 were superior signals for presaging abrupt decline in memory, attention, and executive functioning as compared to blood neurofilament (NfL)(136).
These authors bring forward the proposition that GFAP exemplifies and acts as a proxy for over-energized astrocytes displaying a pro-inflammatory phenotype which are responsible for thwarting neuronal loss secondary to accumulation of Aβ aggregates within the brain tissues(136). Thus, the authors of this study propose putting into practice assaying serum GFAP as an early biomarker for identifying high-risk cases in the incipient stages of brain tissue damage as well as for monitoring disease pathology, progression & prognosis of AD(136).
Blood GFAP [Area under curve (AUC): 0.69-0.96)] had outsmarted CSF GFAP [AUC: 0.59-0.76)] in separating Aβ positive individuals from Aβ negative individuals in AD continuum groups(137). In a observational cross-sectional study in Montreal, Canada (TRIAD: The Translational Biomarkers in Aging and Dementia) the mean [Standard Deviation] values of blood GFAP in cognitive unimpaired (CU) - Aβ-negative group, CU Aβ-positive group, mild cognitive impairment [MCI]-Aβ-positive group and AD group were 185.1 [93.5] pg/mL, 285.0 [142.6] pg/mL, 332.5 [153.6] pg/mL and 388.1 [152.8] pg/mL respectively(137).Blood GFAP was not only accurate in forewarning the transition from MCI status to full-blown AD dementia but also was sensitive to segregate AD from the fronto-temporal dementia (FTD)(138). In this regard,, blood GFAP levels in AD dementia and FTD were (median 375 pg/mL, IQR 276-505 pg/mL) and (190 pg/mL, IQR 134-298 pg/mL, p<0.001) respectively(138). The diagnostic potential of blood GFAP (sensitivity 98% & specificity 60%) in portending AD were much higher than blood phospho-tau (sensitivity 80% & specificity 87%) and serum NfL (neurofilament) (sensitivity 92% & specificity 49%)(138).
In autosomal dominant AD, it was uncovered that blood GFAP elevation happens at least 10 years before symptom onset in the asymptomatic phase of the disease (139). Blood GFAP but not CSF GFAP will anticipate the Aβ accumulation, neurodegeneration, and cognitive decline with more precision and will show an upsurge with a corresponding increase in clinical severity and disease progression in AD patients(121, 139). In study completed by Oeckl, P. et al, blood GFAP was successful in differentiating AD from frontotemporal dementia (FTD) with a higher sensitivity (89%) and specificity (79%)(140). A recent study revealed that, blood GFAP measurements can relay information regarding accumulated Aβ PET burden even in patients with normal CSF Aβ 42/40 levels(121). , Alternatively, blood GFAP levels was also elevated in parallel to the CSF Aβ 42/40 levels in few patients and projected
Aβ PET accumulation in the neocortical regions thereby providing a reliable benchmark for differentiating cognitive unimpaired from cognitively impaired AD patients(121).
What is the relationship between Aβ, phosphor-tau and GFAP in the preclinical and clinical stages of AD? Does serum GFAP accurately predicts brain structural alterations.
Reports suggest that assessing blood GFAP levels is a better plan of action as it surpasses the quantification of blood Aβ-42/1-40 and phosphor-tau181 in portending longitudinal brain Aβ load as well as cognitive deficits in the preclinical and clinical AD patient populations(141, 142). Not only that, calibrating the blood GFAP is even more rewarding because it appraises clinicians regarding the AD related brain structural changes springing up in nascent stages of disease process at a relatively earlier timeframe as compared to utilizing plasma phospho-tau levels(143).
Specifically, it has been regarded as a paramount biomarker for analyzing the role of astrogliosis as well as evaluating the efficacy of therapeutic modalities to arrest the disease progression in AD(121). In a clinical research study by Chatterjee, P. et al it was disclosed that higher blood GFAP levels and lower Aβ1–42/Aβ1–40 ratios were demonstrated to be a harbinger for higher PET Aβ load as well as poorer cognitive performance in the AD patients(144). AD patients exhibiting higher blood GFAP levels and significant Aβ burden were also known to have GFAP-IgG seropositivity thereby underling the presence of silent and dormant autoimmune diseases in these high-risk populations(144). Surprisingly even after adjusting PET Aβ load, blood GFAP positively correlated with progressive cognitive decline in the AD patients. (121, 145).
These findings reinforce the fact that astrocytosis transpiring irrespective of Aβ accumulation will be detrimental in instigating the neuropathological changes in the cerebral cortex and limbic system.
It has been long thought that astrocytes are inherently programmed to defend and safeguard against Aβ and tau accumulation within the neuronal tissuesu(146). Some studies even proved that blood GFAP levels accurately correlated with cumulative Aβ and tau PET burden in the AD patients(147). In contrast, recent studies suggest that blood GFAP accurately predicts the Aβ associated pathology more accurately than tau related neuro-fibrillary tangles (NFT) aggregation(121). In fact, the relationship between blood GFAP and PET-tau burden was nullified after controlling amyloid burden on PET(121). In parallel to these findings, in the study performed by Bellaver, B. et al abnormal reactivity and signaling of astrocytes abutting Aβ was the primordial event that subsequently triggered the spawning of tau aggregates within the preclinical AD groups(148). They surmise that Aβ amassment leads to proliferation of astrocytes which eventually become infuriated leading to discharge of inflammatory mediators such as ROS, cytokines, chemokines, and caspases. (148). These astrocyte released toxic secretions might directly or indirectly enkindle the inception of tau aggregates within the brain tissues in AD(148).
These findings highlight the perception that the accumulation of tau related NFT is a repercussion of astrocytic counter defense to the Aβ aggregation with the neuronal tissues in AD(137, 148). It is conceivable that that GFAP-positive astrocyte populations responding to Aβ and tau aggregates in the AD brain tend to have different genetic makeup for devising discrepant immunological defenses against each of these aggregates. Accordingly, a recent study showed that genetic fingerprint of GFAP-positive astrocyte population associated with Aβ aggregates was predominated with proteostasis and exocytosis genes whereas those juxtaposed to tau aggregates had prevalence of genes associated with DNA/RNA processing, insulin signaling and cytoskeleton dynamics(147). In the same study it was proved that blood GFAP levels are more likely to mirror image the accumulation of with Aβ aggregates rather than tau accumulation in mouse models of AD(147).
Blood GFAP otCSF GFAP in presaging amyloid Aβ PET positivity, cognitive decline and Aβ abetted brain structural alterations(121).
Similarly, blood GFAP outpaced CSF GFAP in foreboding Aβ & tau PET status as well as their respective accumulation status in the brain tissues of preclinical and clinical AD groups(137). Secondly, this study showed that both GFAP and YKL-40 were instrumental in the manifestation of hippocampal atrophy and cognitive impairment in the AD continuum groups(122). 200 patients from Mayo Clinic Study of Aging undergoing PET and MRI imaging were simultaneously investigated for blood GFAP levels(149). They were able to reinforce that GFAP levels were directly proportional to the cortical thickness, white matter hyperintensities (WMH) and lobar cerebral microbleeds (CMB)(149). Blood GFAP levels exhibited a significant positive correlation with white matter atrophy in the temporal & parietal regions and also for memory impairment(150, 151). Excessive Aβ accumulation was shown to provoke grey matter atrophy in the hippocampus, posterior cingulate gyrus, precuneus regions, and temporal lobes(152). These grey matter changes can be monitored by assaying blood GFAP with or without the assistance of PET imaging due to its accuracy in estimating Aβ assemblage.
Pivotal take home messages
Deceleration
Funding
We do not have any financial support for this study.
Conflict of interest
The authors declare no conflict of interest regarding the publication of this paper.
Availability of data and material
The datasets analyzed during the current study are available upon request with no restriction.
Consent for publication
This manuscript has been approved for publication by all authors.
| 1. Chen XQ, Mobley WC. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front Neurosci. 2019;13:659. https://doi.org/10.3389/fnins.2019.00659 PMid:31293377 PMCid:PMC6598402 |
||||
| 2. Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. The Lancet Neurology. 2016;15(7):673-84. https://doi.org/10.1016/S1474-4422(16)00070-3 PMid:27068280 |
||||
| 3. Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, et al. Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Science translational medicine. 2013;5(189):189ra77-ra77. https://doi.org/10.1126/scitranslmed.3005615 PMCid:PMC3838868 |
||||
| 4. Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. The Lancet Neurology. 2003;2(10):605-13. https://doi.org/10.1016/S1474-4422(03)00530-1 PMid:14505582 |
||||
| 5. Kvartsberg H, Portelius E, Andreasson U, Brinkmalm G, Hellwig K, Lelental N, et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer's disease patients and healthy controls. Alzheimer's research & therapy. 2015;7(1):1-9. https://doi.org/10.1186/s13195-015-0124-3 PMid:26136856 PMCid:PMC4487851 |
||||
| 6. Mattsson N, Andreasson U, Zetterberg H, Blennow K, Initiative AsDN. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA neurology. 2017;74(5):557-66. https://doi.org/10.1001/jamaneurol.2016.6117 PMid:28346578 PMCid:PMC5822204 |
||||
| 7. Zou K, Abdullah M, Michikawa M. Current Biomarkers for Alzheimer's Disease: From CSF to Blood. J Pers Med. 2020;10(3). https://doi.org/10.3390/jpm10030085 PMid:32806668 PMCid:PMC7564023 |
||||
| 8. Brinkmalm A, Dumurgier J, Brinkmalm G, Hansson O, Zetterberg H, Bouaziz-Amar E, et al. The pre-synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer's disease. Alzheimer's Research & Therapy. 2016;8(1). https://doi.org/10.1186/s13195-016-0208-8 PMid:27716408 PMCid:PMC5048479 |
||||
| 9. Brinkmalm A, Brinkmalm G, Honer WG, Frölich L, Hausner L, Minthon L, et al. SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer's disease. Molecular neurodegeneration. 2014;9:1-13. https://doi.org/10.1186/1750-1326-9-53 PMid:25418885 PMCid:PMC4253625 |
||||
| 10. Yasuno F, Watanabe A, Kimura Y, Yamauchi Y, Ogata A, Ikenuma H, et al. Estimation of blood-based biomarkers of glial activation related to neuroinflammation. Brain Behav Immun Health. 2022;26:100549. https://doi.org/10.1016/j.bbih.2022.100549 PMid:36388135 PMCid:PMC9650015 |
||||
| 11. Abdullah M, Kimura N, Akatsu H, Hashizume Y, Ferdous T, Tachita T, et al. Flotillin is a novel diagnostic blood marker of Alzheimer's disease. Journal of Alzheimer's Disease. 2019;72(4):1165-76. https://doi.org/10.3233/JAD-190908 PMid:31683489 |
||||
| 12. Abdullah M, Takase H, Nunome M, Enomoto H, Ito J-i, Gong J-S, et al. Amyloid-β reduces exosome release from astrocytes by enhancing JNK phosphorylation. Journal of Alzheimer's Disease. 2016;53(4):1433-41. https://doi.org/10.3233/JAD-160292 PMid:27392863 |
||||
| 13. Nordberg A. Molecular imaging in Alzheimer's disease: new perspectives on biomarkers for early diagnosis and drug development. Alzheimer's Research & Therapy. 2011;3(6):34. https://doi.org/10.1186/alzrt96 PMid:22136152 PMCid:PMC3308023 |
||||
| 14. Perani D, Iaccarino L, Lammertsma AA, Windhorst AD, Edison P, Boellaard R, et al. A new perspective for advanced positron emission tomography-based molecular imaging in neurodegenerative proteinopathies. Alzheimer's & Dementia. 2019;15(8):1081-103. https://doi.org/10.1016/j.jalz.2019.02.004 PMid:31230910 |
||||
| 15. Rodriguez-Vieitez E, Nordberg A. Imaging neuroinflammation: quantification of astrocytosis in a multitracer PET approach. Biomarkers for Alzheimer's Disease Drug Development. 2018:231-51. https://doi.org/10.1007/978-1-4939-7704-8_16 PMid:29512077 |
||||
| 16. Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Molecular Neurodegeneration. 2017;12(1):19. https://doi.org/10.1186/s13024-017-0162-3 PMid:28219440 PMCid:PMC5319037 |
||||
| 17. Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, et al. Evidence for Astrocytosis in Prodromal Alzheimer Disease Provided by 11C-Deuterium-L-Deprenyl: A Multitracer PET Paradigm Combining 11C-Pittsburgh Compound B and 18F-FDG. Journal of Nuclear Medicine. 2012;53(1):37-46. https://doi.org/10.2967/jnumed.110.087031 PMid:22213821 |
||||
| 18. Fowler JS, Logan J, Volkow ND, Wang G-J. Translational Neuroimaging: Positron Emission Tomography Studies of Monoamine Oxidase. Molecular Imaging and Biology. 2005;7(6):377-87. https://doi.org/10.1007/s11307-005-0016-1 PMid:16265597 |
||||
| 19. Kadir A, Marutle A, Gonzalez D, Schöll M, Almkvist O, Mousavi M, et al. Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer's disease. Brain. 2010;134(1):301-17. https://doi.org/10.1093/brain/awq349 PMid:21149866 PMCid:PMC3009843 |
||||
| 20. Ni R, Röjdner J, Voytenko L, Dyrks T, Thiele A, Marutle A, et al. In vitro Characterization of the Regional Binding Distribution of Amyloid PET Tracer Florbetaben and the Glia Tracers Deprenyl and PK11195 in Autopsy Alzheimer's Brain Tissue. Journal of Alzheimer's Disease. 2021;80:1723-37. https://doi.org/10.3233/JAD-201344 PMid:33749648 PMCid:PMC8150513 |
||||
| 21. Kumar A, Fontana IC, Nordberg A. Reactive astrogliosis: A friend or foe in the pathogenesis of Alzheimer's disease. Journal of Neurochemistry. 2023;164(3):309-24. https://doi.org/10.1111/jnc.15565 PMid:34931315 |
||||
| 22. Yang Z, Wang KK. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends in neurosciences. 2015;38(6):364-74. https://doi.org/10.1016/j.tins.2015.04.003 PMid:25975510 PMCid:PMC4559283 |
||||
| 23. Benedet AL, Milà-Alomà M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurology. 2021;78(12):1471-83. https://doi.org/10.1001/jamaneurol.2021.3671 PMid:34661615 PMCid:PMC8524356 |
||||
| 24. Thomsen R, Daugaard TF, Holm IE, Nielsen AL. Alternative mRNA splicing from the glial fibrillary acidic protein (GFAP) gene generates isoforms with distinct subcellular mRNA localization patterns in astrocytes. PLoS One. 2013;8(8):e72110. https://doi.org/10.1371/journal.pone.0072110 PMid:23991052 PMCid:PMC3753360 |
||||
| 25. Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One. 2012;7(8):e42823. https://doi.org/10.1371/journal.pone.0042823 PMid:22912745 PMCid:PMC3418292 |
||||
| 26. Potokar M, Morita M, Wiche G, Jorgačevski J. The Diversity of Intermediate Filaments in Astrocytes. Cells. 2020;9(7). https://doi.org/10.3390/cells9071604 PMid:32630739 PMCid:PMC7408014 |
||||
| 27. Lin N-H, Yang A-W, Chang C-H, Perng M-D. Elevated GFAP isoform expression promotes protein aggregation and compromises astrocyte function. The FASEB Journal. 2021;35(5):e21614. https://doi.org/10.1096/fj.202100087R |
||||
| 28. Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol. 2011;93(3):421-43. https://doi.org/10.1016/j.pneurobio.2011.01.005 PMid:21219963 |
||||
| 29. Cho W, Messing A. Properties of astrocytes cultured from GFAP over-expressing and GFAP mutant mice. Experimental Cell Research. 2009;315(7):1260-72. https://doi.org/10.1016/j.yexcr.2008.12.012 PMid:19146851 PMCid:PMC2665202 |
||||
| 30. Stassen O, van Bodegraven EJ, Giuliani F, Moeton M, Kanski R, Sluijs JA, et al. GFAPδ/GFAPα ratio directs astrocytoma gene expression towards a more malignant profile. Oncotarget. 2017;8(50):88104-21. https://doi.org/10.18632/oncotarget.21540 PMid:29152145 PMCid:PMC5675697 |
||||
| 31. van Bodegraven EJ, van Asperen JV, Sluijs JA, van Deursen CBJ, van Strien ME, Stassen O, et al. GFAP alternative splicing regulates glioma cell-ECM interaction in a DUSP4-dependent manner. Faseb j. 2019;33(11):12941-59. https://doi.org/10.1096/fj.201900916R PMid:31480854 |
||||
| 32. Elobeid A, Bongcam-Rudloff E, Westermark B, Nistér M. Effects of inducible glial fibrillary acidic protein on glioma cell motility and proliferation. J Neurosci Res. 2000;60(2):245-56. https://doi.org/10.1002/(SICI)1097-4547(20000415)60:2<245::AID-JNR14>3.0.CO;2-1 |
||||
| 33. Uceda-Castro R, van Asperen JV, Vennin C, Sluijs JA, van Bodegraven EJ, Margarido AS, et al. GFAP splice variants fine-tune glioma cell invasion and tumour dynamics by modulating migration persistence. Scientific Reports. 2022;12(1):424. https://doi.org/10.1038/s41598-021-04127-5 PMid:35013418 PMCid:PMC8748899 |
||||
| 34. van Asperen JV, Robe P, Hol EM. GFAP Alternative Splicing and the Relevance for Disease - A Focus on Diffuse Gliomas. ASN Neuro. 2022;14:17590914221102065. https://doi.org/10.1177/17590914221102065 PMid:35673702 PMCid:PMC9185002 |
||||
| 35. Moeton M, Kanski R, Stassen OM, Sluijs JA, Geerts D, van Tijn P, et al. Silencing GFAP isoforms in astrocytoma cells disturbs laminin-dependent motility and cell adhesion. Faseb j. 2014;28(7):2942-54. https://doi.org/10.1096/fj.13-245837 PMid:24696300 |
||||
| 36. Radu R, Petrescu GED, Gorgan RM, Brehar FM. GFAPδ: A Promising Biomarker and Therapeutic Target in Glioblastoma. Front Oncol. 2022;12:859247. https://doi.org/10.3389/fonc.2022.859247 PMid:35372061 PMCid:PMC8971704 |
||||
| 37. Perng MD, Wen SF, Gibbon T, Middeldorp J, Sluijs J, Hol EM, et al. Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-delta, but with consequences for filament organization and alphaB-crystallin association. Mol Biol Cell. 2008;19(10):4521-33. https://doi.org/10.1091/mbc.e08-03-0284 PMid:18685083 PMCid:PMC2555932 |
||||
| 38. Moeton M, Stassen OM, Sluijs JA, van der Meer VW, Kluivers LJ, van Hoorn H, et al. GFAP isoforms control intermediate filament network dynamics, cell morphology, and focal adhesions. Cell Mol Life Sci. 2016;73(21):4101-20. https://doi.org/10.1007/s00018-016-2239-5 PMid:27141937 PMCid:PMC5043008 |
||||
| 39. Bugiani M, Boor I, van Kollenburg B, Postma N, Polder E, van Berkel C, et al. Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol. 2011;70(1):69-82. https://doi.org/10.1097/NEN.0b013e318203ae74 PMid:21157376 PMCid:PMC4135437 |
||||
| 40. Roelofs RF, Fischer DF, Houtman SH, Sluijs JA, Van Haren W, Van Leeuwen FW, et al. Adult human subventricular, subgranular, and subpial zones contain astrocytes with a specialized intermediate filament cytoskeleton. Glia. 2005;52(4):289-300. https://doi.org/10.1002/glia.20243 PMid:16001427 |
||||
| 41. van den Berge SA, Middeldorp J, Zhang CE, Curtis MA, Leonard BW, Mastroeni D, et al. Longterm quiescent cells in the aged human subventricular neurogenic system specifically express GFAP-delta. Aging Cell. 2010;9(3):313-26. https://doi.org/10.1111/j.1474-9726.2010.00556.x PMid:20121722 |
||||
| 42. Leonard BW, Mastroeni D, Grover A, Liu Q, Yang K, Gao M, et al. Subventricular zone neural progenitors from rapid brain autopsies of elderly subjects with and without neurodegenerative disease. J Comp Neurol. 2009;515(3):269-94. https://doi.org/10.1002/cne.22091 https://doi.org/10.1002/cne.22040 PMid:19425077 PMCid:PMC2757160 |
||||
| 43. Middeldorp J, Boer K, Sluijs JA, De Filippis L, Encha-Razavi F, Vescovi AL, et al. GFAPdelta in radial glia and subventricular zone progenitors in the developing human cortex. Development. 2010;137(2):313-21. https://doi.org/10.1242/dev.041632 PMid:20040497 |
||||
| 44. Kamphuis W, Middeldorp J, Kooijman L, Sluijs JA, Kooi E-J, Moeton M, et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiology of Aging. 2014;35(3):492-510. https://doi.org/10.1016/j.neurobiolaging.2013.09.035 PMid:24269023 |
||||
| 45. Heo DH, Kim SH, Yang KM, Cho YJ, Kim KN, Yoon DH, et al. A histopathological diagnostic marker for human spinal astrocytoma: expression of glial fibrillary acidic protein-δ. J Neurooncol. 2012;108(1):45-52. https://doi.org/10.1007/s11060-012-0801-z PMid:22318658 |
||||
| 46. Martí-Fàbregas J, Romaguera-Ros M, Gómez-Pinedo U, Martínez-Ramírez S, Jiménez-Xarrié E, Marín R, et al. Proliferation in the human ipsilateral subventricular zone after ischemic stroke. Neurology. 2010;74(5):357-65. https://doi.org/10.1212/WNL.0b013e3181cbccec PMid:20054008 |
||||
| 47. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. Febs j. 2011;278(1):16-27. https://doi.org/10.1111/j.1742-4658.2010.07919.x PMid:21087457 |
||||
| 48. Reich R, Blumenthal M, Liscovitch M. Role of phospholipase D in laminin-induced production of gelatinase A (MMP-2) in metastatic cells. Clin Exp Metastasis. 1995;13(2):134-40. https://doi.org/10.1007/BF00133618 PMid:7882615 |
||||
| 49. Nakagawa T, Kubota T, Kabuto M, Fujimoto N, Okada Y. Secretion of matrix metalloproteinase-2 (72 kD gelatinase/type IV collagenase = gelatinase A) by malignant human glioma cell lines: implications for the growth and cellular invasion of the extracellular matrix. J Neurooncol. 1996;28(1):13-24. https://doi.org/10.1007/BF00300442 PMid:8740587 |
||||
| 50. Middeldorp J, van den Berge SA, Aronica E, Speijer D, Hol EM. Specific human astrocyte subtype revealed by affinity purified GFAP antibody; unpurified serum cross-reacts with neurofilament-L in Alzheimer. PLoS One. 2009;4(11):e7663. https://doi.org/10.1371/journal.pone.0007663 PMid:19888461 PMCid:PMC2766629 |
||||
| 51. Hol EM, Roelofs RF, Moraal E, Sonnemans MAF, Sluijs JA, Proper EA, et al. Neuronal expression of GFAP in patients with Alzheimer pathology and identification of novel GFAP splice forms. Molecular Psychiatry. 2003;8(9):786-96. https://doi.org/10.1038/sj.mp.4001379 PMid:12931206 |
||||
| 52. Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144(1):16-26. https://doi.org/10.1016/j.cell.2010.11.056 PMid:21215366 PMCid:PMC3038581 |
||||
| 53. Boer K, Middeldorp J, Spliet WG, Razavi F, van Rijen PC, Baayen JC, et al. Immunohistochemical characterization of the out-of frame splice variants GFAP Delta164/Deltaexon 6 in focal lesions associated with chronic epilepsy. Epilepsy Res. 2010;90(1-2):99-109. https://doi.org/10.1016/j.eplepsyres.2010.03.014 PMid:20430588 |
||||
| 54. Martinian L, Boer K, Middeldorp J, Hol EM, Sisodiya SM, Squier W, et al. Expression patterns of glial fibrillary acidic protein (GFAP)-delta in epilepsy-associated lesional pathologies. Neuropathology and Applied Neurobiology. 2009;35(4):394-405. https://doi.org/10.1111/j.1365-2990.2008.00996.x PMid:19508443 |
||||
| 55. Oberheim NA, Takano T, Han X, He W, Lin JHC, Wang F, et al. Uniquely Hominid Features of Adult Human Astrocytes. The Journal of Neuroscience. 2009;29(10):3276-87. https://doi.org/10.1523/JNEUROSCI.4707-08.2009 PMid:19279265 PMCid:PMC2819812 |
||||
| 56. Zelenika D, Grima B, Brenner M, Pessac B. A novel glial fibrillary acidic protein mRNA lacking exon 1. Brain Res Mol Brain Res. 1995;30(2):251-8. https://doi.org/10.1016/0169-328X(95)00010-P PMid:7637576 |
||||
| 57. Blechingberg J, Holm IE, Nielsen KB, Jensen TH, Jørgensen AL, Nielsen AL. Identification and characterization of GFAPkappa, a novel glial fibrillary acidic protein isoform. Glia. 2007;55(5):497-507. https://doi.org/10.1002/glia.20475 PMid:17203480 |
||||
| 58. Middeldorp J, Kamphuis W, Sluijs JA, Achoui D, Leenaars CH, Feenstra MG, et al. Intermediate filament transcription in astrocytes is repressed by proteasome inhibition. Faseb j. 2009;23(8):2710-26. https://doi.org/10.1096/fj.08-127696 PMid:19332645 PMCid:PMC3221645 |
||||
| 59. Nielsen AL, Jørgensen AL. Self-assembly of the cytoskeletal glial fibrillary acidic protein is inhibited by an isoform-specific C terminus. J Biol Chem. 2004;279(40):41537-45. https://doi.org/10.1074/jbc.M406601200 PMid:15284230 |
||||
| 60. Chen W-J, Liem R. The endless story of the glial fibrillary acidic protein. Journal of Cell Science. 1994;107(8):2299-311. https://doi.org/10.1242/jcs.107.8.2299 PMid:7983188 |
||||
| 61. Condorelli DF, Nicoletti VG, Dell'Albani P, Barresi V, Caruso A, Conticello SG, et al. GFAPbeta mRNA expression in the normal rat brain and after neuronal injury. Neurochem Res. 1999;24(5):709-14. https://doi.org/10.1023/A:1021016828704 PMid:10344602 |
||||
| 62. Galea E, Dupouey P, Feinstein DL. Glial fibrillary acidic protein mRNA isotypes: expression in vitro and in vivo. J Neurosci Res. 1995;41(4):452-61. https://doi.org/10.1002/jnr.490410404 PMid:7473876 |
||||
| 63. Benveniste EN, Vidovic M, Panek RB, Norris JG, Reddy AT, Benos DJ. Interferon-gamma-induced astrocyte class II major histocompatibility complex gene expression is associated with both protein kinase C activation and Na+ entry. J Biol Chem. 1991;266(27):18119-26. https://doi.org/10.1016/S0021-9258(18)55244-3 PMid:1917946 |
||||
| 64. Stevens B. Glia: much more than the neuron's side-kick. Current Biology. 2003;13(12):R469-R72. https://doi.org/10.1016/S0960-9822(03)00404-4 PMid:12814563 |
||||
| 65. Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453-7. https://doi.org/10.1038/nm838 PMid:12612547 |
||||
| 66. Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2016;275 Pt 3(0 3):305-15. https://doi.org/10.1016/j.expneurol.2015.03.020 PMid:25828533 PMCid:PMC4586307 |
||||
| 67. Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. 2014;7(2):a020420. https://doi.org/10.1101/cshperspect.a020420 PMid:25380660 PMCid:PMC4315924 |
||||
| 68. Kim H, Lee EJ, Lim YM, Kim KK. Glial Fibrillary Acidic Protein in Blood as a Disease Biomarker of Neuromyelitis Optica Spectrum Disorders. Front Neurol. 2022;13:865730. https://doi.org/10.3389/fneur.2022.865730 PMid:35370870 PMCid:PMC8968934 |
||||
| 69. Crols R, Saerens J, Noppe M, Lowenthal A. Increased GFAp levels in CSF as a marker of organicity in patients with Alzheimer's disease and other types of irreversible chronic organic brain syndrome. J Neurol. 1986;233(3):157-60. https://doi.org/10.1007/BF00314423 PMid:3522811 |
||||
| 70. Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2(4):492-516. https://doi.org/10.1007/s12975-011-0125-x PMid:22299022 PMCid:PMC3268209 |
||||
| 71. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584-96. https://doi.org/10.1038/nm.3407 PMid:24309662 PMCid:PMC4080800 |
||||
| 72. Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci. 2015;35(2):518-26. https://doi.org/10.1523/JNEUROSCI.3742-14.2015 PMid:25589747 PMCid:PMC4293408 |
||||
| 73. Mestre H, Mori Y, Nedergaard M. The Brain's Glymphatic System: Current Controversies. Trends Neurosci. 2020;43(7):458-66. https://doi.org/10.1016/j.tins.2020.04.003 PMid:32423764 PMCid:PMC7331945 |
||||
| 74. Da Mesquita S, Fu Z, Kipnis J. The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron. 2018;100(2):375-88. https://doi.org/10.1016/j.neuron.2018.09.022 PMid:30359603 PMCid:PMC6268162 |
||||
| 75. Sans M, Panés J, Ardite E, Elizalde JI, Arce Y, Elena M, et al. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology. 1999;116(4):874-83. https://doi.org/10.1016/S0016-5085(99)70070-3 PMid:10092309 |
||||
| 76. Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer's disease. Neurobiol Dis. 2017;107:41-56. https://doi.org/10.1016/j.nbd.2016.07.007 PMid:27425887 PMCid:PMC5600438 |
||||
| 77. Rahmouni N, Tissot C, Lussier FZ, Bezgin G, Therriault J, Stevenson J, et al. Associations between neutrophils and amyloid deposition in the Alzheimer's disease spectrum. Alzheimer's & Dementia. 2021;17(S1):e056652. https://doi.org/10.1002/alz.056652 |
||||
| 78. Azevedo EP, Guimarães-Costa AB, Torezani GS, Braga CA, Palhano FL, Kelly JW, et al. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J Biol Chem. 2012;287(44):37206-18. https://doi.org/10.1074/jbc.M112.369942 PMid:22918834 PMCid:PMC3481320 |
||||
| 79. Zaghi J, Goldenson B, Inayathullah M, Lossinsky AS, Masoumi A, Avagyan H, et al. Alzheimer disease macrophages shuttle amyloid-beta from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathol. 2009;117(2):111-24. https://doi.org/10.1007/s00401-008-0481-0 PMid:19139910 PMCid:PMC5650912 |
||||
| 80. Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta. 2016;1862(5):1037-46. https://doi.org/10.1016/j.bbadis.2015.08.024 PMid:26327684 PMCid:PMC4827375 |
||||
| 81. Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R, et al. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell. 2013;12(2):224-36. https://doi.org/10.1111/acel.12045 PMid:23413811 |
||||
| 82. Gireud-Goss M, Mack AF, McCullough LD, Urayama A. Cerebral Amyloid Angiopathy and Blood-Brain Barrier Dysfunction. Neuroscientist. 2021;27(6):668-84. https://doi.org/10.1177/1073858420954811 PMid:33238806 PMCid:PMC9853919 |
||||
| 83. Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010;14(5):1101-12. https://doi.org/10.1111/j.1582-4934.2009.00717.x PMid:19438816 PMCid:PMC3822747 |
||||
| 84. Marco S, Skaper SD. Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401(3):219-24. https://doi.org/10.1016/j.neulet.2006.03.047 PMid:16644119 |
||||
| 85. Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15(5):1167-78. https://doi.org/10.1089/ars.2011.3895 PMid:21294650 |
||||
| 86. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106(12):1489-99. https://doi.org/10.1172/JCI10498 PMid:11120756 PMCid:PMC387254 |
||||
| 87. Elali A, Rivest S. The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer's disease. Front Physiol. 2013;4:45. https://doi.org/10.3389/fphys.2013.00045 PMid:23494712 PMCid:PMC3595512 |
||||
| 88. Ito S, Ohtsuki S, Murata S, Katsukura Y, Suzuki H, Funaki M, et al. Involvement of insulin-degrading enzyme in insulin- and atrial natriuretic peptide-sensitive internalization of amyloid-β peptide in mouse brain capillary endothelial cells. J Alzheimers Dis. 2014;38(1):185-200. https://doi.org/10.3233/JAD-122077 PMid:23948938 |
||||
| 89. Preston SD, Steart PV, Wilkinson A, Nicoll JAR, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathology and Applied Neurobiology. 2003;29(2):106-17. https://doi.org/10.1046/j.1365-2990.2003.00424.x PMid:12662319 |
||||
| 90. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12(12):723-38. https://doi.org/10.1038/nrn3114 PMid:22048062 PMCid:PMC4036520 |
||||
| 91. Pahnke J, Langer O, Krohn M. Alzheimer's and ABC transporters--new opportunities for diagnostics and treatment. Neurobiol Dis. 2014;72 Pt A:54-60. https://doi.org/10.1016/j.nbd.2014.04.001 PMid:24746857 PMCid:PMC4199932 |
||||
| 92. Sturmey E, Malaspina A. Blood biomarkers in ALS: challenges, applications and novel frontiers. Acta Neurologica Scandinavica. 2022;146(4):375-88. https://doi.org/10.1111/ane.13698 PMid:36156207 PMCid:PMC9828487 |
||||
| 93. Nimmo J, Johnston DA, Dodart JC, MacGregor-Sharp MT, Weller RO, Nicoll JAR, et al. Peri-arterial pathways for clearance of α-Synuclein and tau from the brain: Implications for the pathogenesis of dementias and for immunotherapy. Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring. 2020;12(1):e12070. https://doi.org/10.1002/dad2.12070 PMid:32782922 PMCid:PMC7409108 |
||||
| 94. van Veluw SJ, Hou SS, Calvo-Rodriguez M, Arbel-Ornath M, Snyder AC, Frosch MP, et al. Vasomotion as a Driving Force for Paravascular Clearance in the Awake Mouse Brain. Neuron. 2020;105(3):549-61.e5. https://doi.org/10.1016/j.neuron.2019.10.033 PMid:31810839 PMCid:PMC7028316 |
||||
| 95. Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008;18(2):253-66. https://doi.org/10.1111/j.1750-3639.2008.00133.x PMid:18363936 PMCid:PMC8095597 |
||||
| 96. Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The Glymphatic System: A Beginner's Guide. Neurochem Res. 2015;40(12):2583-99. https://doi.org/10.1007/s11064-015-1581-6 PMid:25947369 PMCid:PMC4636982 |
||||
| 97. Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30-42. https://doi.org/10.1038/s41582-019-0281-2 PMid:31827267 PMCid:PMC7268202 |
||||
| 98. Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457-70. https://doi.org/10.1038/nrneurol.2015.119 PMid:26195256 PMCid:PMC4694579 |
||||
| 99. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373-7. https://doi.org/10.1126/science.1241224 PMid:24136970 PMCid:PMC3880190 |
||||
| 100. Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34(2):131-44. https://doi.org/10.1111/j.1365-2990.2007.00926.x PMid:18208483 |
||||
| 101. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4(147):147ra11. https://doi.org/10.1126/scitranslmed.3003748 PMid:22896675 PMCid:PMC3551275 |
||||
| 102. Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33(46):18190-9. https://doi.org/10.1523/JNEUROSCI.1592-13.2013 PMid:24227727 PMCid:PMC3866416 |
||||
| 103. Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136(1):139-52. https://doi.org/10.1007/s00401-018-1862-7 PMid:29754206 PMCid:PMC6015107 |
||||
| 104. Iliff JJ, Nedergaard M. Is there a cerebral lymphatic system? Stroke. 2013;44(6 Suppl 1):S93-5. https://doi.org/10.1161/STROKEAHA.112.678698 PMid:23709744 PMCid:PMC3699410 |
||||
| 105. Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76(6):845-61. https://doi.org/10.1002/ana.24271 PMid:25204284 PMCid:PMC4245362 |
||||
| 106. Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, et al. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. 2013;123(3):1299-309. https://doi.org/10.1172/JCI67677 PMid:23434588 PMCid:PMC3582150 |
||||
| 107. Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO. Disruption of arterial perivascular drainage of amyloid-β from the brains of mice expressing the human APOE ε4 allele. PLoS One. 2012;7(7):e41636. https://doi.org/10.1371/journal.pone.0041636 PMid:22848551 PMCid:PMC3404985 |
||||
| 108. Nimmo J, Johnston DA, Dodart JC, MacGregor-Sharp MT, Weller RO, Nicoll JAR, et al. Peri-arterial pathways for clearance of α-Synuclein and tau from the brain: Implications for the pathogenesis of dementias and for immunotherapy. Alzheimers Dement (Amst). 2020;12(1):e12070. https://doi.org/10.1002/dad2.12070 PMid:32782922 PMCid:PMC7409108 |
||||
| 109. Hawkes CA, Härtig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121(4):431-43. https://doi.org/10.1007/s00401-011-0801-7 PMid:21259015 |
||||
| 110. Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R, et al. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell. 2013;12(2):224-36. https://doi.org/10.1111/acel.12045 PMid:23413811 |
||||
| 111. Kapaki E, Vakrakou AG, Boufidou F. Novel CSF Biomarkers Tracking Autoimmune Inflammatory and Neurodegenerative Aspects of CNS Diseases. Diagnostics (Basel). 2022;13(1). https://doi.org/10.3390/diagnostics13010073 PMid:36611365 PMCid:PMC9818715 |
||||
| 112. Fukuyama R, Izumoto T, Fushiki S. The Cerebrospinal Fluid Level of Glial Fibrillary Acidic Protein Is Increased in Cerebrospinal Fluid from Alzheimer's Disease Patients and Correlates with Severity of Dementia. European Neurology. 2001;46(1):35-8. https://doi.org/10.1159/000050753 PMid:11455181 |
||||
| 113. Jesse S, Steinacker P, Cepek L, von Arnim CA, Tumani H, Lehnert S, et al. Glial fibrillary acidic protein and protein S-100B: different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer's disease and Creutzfeldt-Jakob disease. J Alzheimers Dis. 2009;17(3):541-51. https://doi.org/10.3233/JAD-2009-1075 PMid:19433893 |
||||
| 114. Abu-Rumeileh S, Steinacker P, Polischi B, Mammana A, Bartoletti-Stella A, Oeckl P, et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res Ther. 2019;12(1):2. https://doi.org/10.1186/s13195-019-0562-4 PMid:31892365 PMCid:PMC6937795 |
||||
| 115. Sun M, Liu N, Xie Q, Li X, Sun J, Wang H, et al. A candidate biomarker of glial fibrillary acidic protein in CSF and blood in differentiating multiple sclerosis and its subtypes: A systematic review and meta-analysis. Mult Scler Relat Disord. 2021;51:102870. https://doi.org/10.1016/j.msard.2021.102870 PMid:33819724 |
||||
| 116. Axelsson M, Malmeström C, Nilsson S, Haghighi S, Rosengren L, Lycke J. Glial fibrillary acidic protein: a potential biomarker for progression in multiple sclerosis. J Neurol. 2011;258(5):882-8. https://doi.org/10.1007/s00415-010-5863-2 PMid:21197541 |
||||
| 117. Petzold A, Eikelenboom MJ, Gveric D, Keir G, Chapman M, Lazeron RH, et al. Markers for different glial cell responses in multiple sclerosis: clinical and pathological correlations. Brain. 2002;125(Pt 7):1462-73. https://doi.org/10.1093/brain/awf165 PMid:12076997 |
||||
| 118. Jany PL, Agosta GE, Benko WS, Eickhoff JC, Keller SR, Köehler W, et al. CSF and Blood Levels of GFAP in Alexander Disease. eneuro. 2015;2(5):ENEURO.0080-15.2015. https://doi.org/10.1523/ENEURO.0080-15.2015 PMid:26478912 PMCid:PMC4603256 |
||||
| 119. Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, et al. Biomarkers of Traumatic Injury Are Transported from Brain to Blood via the Glymphatic System. The Journal of Neuroscience. 2015;35(2):518-26. https://doi.org/10.1523/JNEUROSCI.3742-14.2015 PMid:25589747 PMCid:PMC4293408 |
||||
| 120. Montoliu-Gaya L, Alcolea D, Ashton NJ, Pegueroles J, Levin J, Bosch B, et al. Plasma and cerebrospinal fluid glial fibrillary acidic protein levels in adults with Down syndrome: a longitudinal cohort study. EBioMedicine. 2023;90:104547. https://doi.org/10.1016/j.ebiom.2023.104547 PMid:37002988 PMCid:PMC10070083 |
||||
| 121. Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE, et al. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505-16. https://doi.org/10.1093/brain/awab223 PMid:34259835 PMCid:PMC8677538 |
||||
| 122. Ferrari-Souza JP, Ferreira PCL, Bellaver B, Tissot C, Wang Y-T, Leffa DT, et al. Astrocyte biomarker signatures of amyloid-β and tau pathologies in Alzheimer's disease. Molecular Psychiatry. 2022;27(11):4781-9. https://doi.org/10.1038/s41380-022-01716-2 PMid:35948658 PMCid:PMC9734046 |
||||
| 123. Simrén J, Weninger H, Brum WS, Khalil S, Benedet AL, Blennow K, et al. Differences between blood and cerebrospinal fluid glial fibrillary Acidic protein levels: The effect of sample stability. Alzheimers Dement. 2022;18(10):1988-92. https://doi.org/10.1002/alz.12806 PMid:36102852 PMCid:PMC9826213 |
||||
| 124. Ashton NJ, Suárez-Calvet M, Karikari TK, Lantero-Rodriguez J, Snellman A, Sauer M, et al. Effects of pre-analytical procedures on blood biomarkers for Alzheimer's pathophysiology, glial activation, and neurodegeneration. Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring. 2021;13(1):e12168. https://doi.org/10.1002/dad2.12168 PMid:34124336 PMCid:PMC8171159 |
||||
| 125. Mouser PE, Head E, Ha KH, Rohn TT. Caspase-mediated cleavage of glial fibrillary acidic protein within degenerating astrocytes of the Alzheimer's disease brain. Am J Pathol. 2006;168(3):936-46. https://doi.org/10.2353/ajpath.2006.050798 PMid:16507909 PMCid:PMC1606516 |
||||
| 126. Zoltewicz JS, Scharf D, Yang B, Chawla A, Newsom KJ, Fang L. Characterization of Antibodies that Detect Human GFAP after Traumatic Brain Injury. Biomark Insights. 2012;7:71-9. https://doi.org/10.4137/BMI.S9873 PMid:22798722 PMCid:PMC3394595 |
||||
| 127. Yang Z, Arja RD, Zhu T, Sarkis GA, Patterson RL, Romo P, et al. Characterization of Calpain and Caspase-6-Generated Glial Fibrillary Acidic Protein Breakdown Products Following Traumatic Brain Injury and Astroglial Cell Injury. Int J Mol Sci. 2022;23(16). https://doi.org/10.3390/ijms23168960 PMid:36012232 PMCid:PMC9409281 |
||||
| 128. Simrén J, Weninger H, Brum WS, Khalil S, Benedet AL, Blennow K, et al. Differences between blood and cerebrospinal fluid glial fibrillary Acidic protein levels: The effect of sample stability. Alzheimer's & Dementia. 2022;18(10):1988-92. https://doi.org/10.1002/alz.12806 PMid:36102852 PMCid:PMC9826213 |
||||
| 129. Eide PK, Lashkarivand A, Pripp A, Valnes LM, Hovd MH, Ringstad G, et al. Plasma neurodegeneration biomarker concentrations associate with glymphatic and meningeal lymphatic measures in neurological disorders. Nat Commun. 2023;14(1):2084. https://doi.org/10.1038/s41467-023-37685-5 PMid:37045847 PMCid:PMC10097687 |
||||
| 130. Amalia L. Glial Fibrillary Acidic Protein (GFAP): Neuroinflammation Biomarker in Acute Ischemic Stroke. J Inflamm Res. 2021;14:7501-6. https://doi.org/10.2147/JIR.S342097 PMid:35002283 PMCid:PMC8722682 |
||||
| 131. Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: A matter of blood-brain barrier dysfunction? J Exp Med. 2017;214(11):3151-69. https://doi.org/10.1084/jem.20171406 PMid:29061693 PMCid:PMC5679168 |
||||
| 132. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews Neurology. 2018;14(3):133-50. https://doi.org/10.1038/nrneurol.2017.188 PMid:29377008 PMCid:PMC5829048 |
||||
| 133. Takemura M, Gomi H, Colucci-Guyon E, Itohara S. Protective Role of Phosphorylation in Turnover of Glial Fibrillary Acidic Protein in Mice. The Journal of Neuroscience. 2002;22(16):6972-9. https://doi.org/10.1523/JNEUROSCI.22-16-06972.2002 PMid:12177195 PMCid:PMC6757867 |
||||
| 134. Cicognola C, Janelidze S, Hertze J, Zetterberg H, Blennow K, Mattsson-Carlgren N, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer's Research & Therapy. 2021;13(1):68. https://doi.org/10.1186/s13195-021-00804-9 PMid:33773595 PMCid:PMC8005231 |
||||
| 135. Rajan KB, Aggarwal NT, McAninch EA, Weuve J, Barnes LL, Wilson RS, et al. Remote Blood Biomarkers of Longitudinal Cognitive Outcomes in a Population Study. Ann Neurol. 2020;88(6):1065-76. https://doi.org/10.1002/ana.25874 PMid:32799383 PMCid:PMC9186023 |
||||
| 136. Verberk IMW, Laarhuis MB, van den Bosch KA, Ebenau JL, van Leeuwenstijn M, Prins ND, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic-based cohort study. Lancet Healthy Longev. 2021;2(2):e87-e95. https://doi.org/10.1016/S2666-7568(20)30061-1 PMid:36098162 |
||||
| 137. Benedet AL, Milà-Alomà M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurol. 2021;78(12):1471-83. https://doi.org/10.1001/jamaneurol.2021.3671 PMid:34661615 PMCid:PMC8524356 |
||||
| 138. Oeckl P, Anderl-Straub S, Von Arnim CAF, Baldeiras I, Diehl-Schmid J, Grimmer T, et al. Serum GFAP differentiates Alzheimer's disease from frontotemporal dementia and predicts MCI-to-dementia conversion. J Neurol Neurosurg Psychiatry. 2022. https://doi.org/10.1136/jnnp-2021-328547 PMid:35477892 |
||||
| 139. Chatterjee P, Vermunt L, Gordon BA, Pedrini S, Boonkamp L, Armstrong NJ, et al. Plasma glial fibrillary acidic protein in autosomal dominant Alzheimer's disease: Associations with Aβ-PET, neurodegeneration, and cognition. Alzheimer's & Dementia.n/a(n/a). | ||||
| 140. Oeckl P, Halbgebauer S, Anderl-Straub S, Steinacker P, Huss AM, Neugebauer H, et al. Glial Fibrillary Acidic Protein in Serum is Increased in Alzheimer's Disease and Correlates with Cognitive Impairment. J Alzheimers Dis. 2019;67(2):481-8. https://doi.org/10.3233/JAD-180325 PMid:30594925 |
||||
| 141. Verberk IMW, Thijssen E, Koelewijn J, Mauroo K, Vanbrabant J, de Wilde A, et al. Combination of plasma amyloid beta((1-42/1-40)) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res Ther. 2020;12(1):118. https://doi.org/10.1186/s13195-020-00682-7 PMid:32988409 PMCid:PMC7523295 |
||||
| 142. Chatterjee P, Pedrini S, Ashton NJ, Tegg M, Goozee K, Singh AK, et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer's disease. Alzheimers Dement. 2022;18(6):1141-54. https://doi.org/10.1002/alz.12447 PMid:34494715 |
||||
| 143. Johansson C, Thordardottir S, Laffita-Mesa J, Rodriguez-Vieitez E, Zetterberg H, Blennow K, et al. Plasma biomarker profiles in autosomal dominant Alzheimer's disease. Brain. 2023;146(3):1132-40. https://doi.org/10.1093/brain/awac399 PMid:36626935 PMCid:PMC9976964 |
||||
| 144. Chatterjee P, Pedrini S, Stoops E, Goozee K, Villemagne VL, Asih PR, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Translational Psychiatry. 2021;11(1):27. https://doi.org/10.1038/s41398-020-01137-1 PMid:33431793 PMCid:PMC7801513 |
||||
| 145. Verberk IMW, Laarhuis MB, van den Bosch KA, Ebenau JL, van Leeuwenstijn M, Prins ND, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic-based cohort study. The Lancet Healthy Longevity. 2021;2(2):e87-e95. https://doi.org/10.1016/S2666-7568(20)30061-1 PMid:36098162 |
||||
| 146. Serrano-Pozo A, Mielke ML, Gómez-Isla T, Betensky RA, Growdon JH, Frosch MP, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol. 2011;179(3):1373-84. https://doi.org/10.1016/j.ajpath.2011.05.047 PMid:21777559 PMCid:PMC3157187 |
||||
| 147. Bastiani MAD, Bellaver B, Brum WS, Souza DG, Ferreira PCL, Rocha AS, et al. Hippocampal GFAP-positive astrocyte responses to amyloid and tau pathologies. bioRxiv. 2022:2022.02.25.481812. https://doi.org/10.1101/2022.02.25.481812 |
||||
| 148. Bellaver B, Povala G, Ferreira PCL, Ferrari-Souza JP, Leffa DT, Lussier FZ, et al. Astrocyte reactivity influences amyloid-β effects on tau pathology in preclinical Alzheimer's disease. Nature Medicine. 2023. https://doi.org/10.1038/s41591-023-02380-x PMid:37248300 PMCid:PMC10353939 |
||||
| 149. Shir D, Graff-Radford J, Hofrenning EI, Lesnick TG, Przybelski SA, Lowe VJ, et al. Association of plasma glial fibrillary acidic protein (GFAP) with neuroimaging of Alzheimer's disease and vascular pathology. Alzheimers Dement (Amst). 2022;14(1):e12291. https://doi.org/10.1002/dad2.12291 PMid:35252538 PMCid:PMC8883441 |
||||
| 150. Asken BM, VandeVrede L, Rojas JC, Fonseca C, Staffaroni AM, Elahi FM, et al. Lower White Matter Volume and Worse Executive Functioning Reflected in Higher Levels of Plasma GFAP among Older Adults with and Without Cognitive Impairment. J Int Neuropsychol Soc. 2022;28(6):588-99. https://doi.org/10.1017/S1355617721000813 PMid:34158138 PMCid:PMC8692495 |
||||
| 151. Knopman DS, Lundt ES, Therneau TM, Vemuri P, Lowe VJ, Kantarci K, et al. Entorhinal cortex tau, amyloid-β, cortical thickness and memory performance in non-demented subjects. Brain. 2019;142(4):1148-60. https://doi.org/10.1093/brain/awz025 PMid:30759182 PMCid:PMC6439321 |
||||
| 152. Doré V, Villemagne VL, Bourgeat P, Fripp J, Acosta O, Chetélat G, et al. Cross-sectional and longitudinal analysis of the relationship between Aβ deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurol. 2013;70(7):903-11. https://doi.org/10.1001/jamaneurol.2013.1062 PMid:23712469 |
||||

| Article View | 1,048 |
| PDF Download | 624 |